Statistical modeling in pharmaceutical research and development.
Diseases of carbohydrate_metabolism
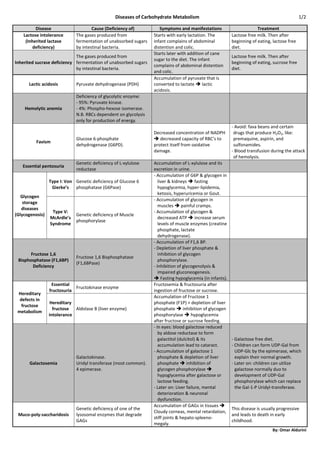
1. Diseases of Carbohydrate Metabolism 1/2
Disease Cause (Deficiency of) Symptoms and manifestations Treatment
Lactose intolerance The gases produced from Starts with early lactation. The Lactose free milk. Then after
(Inherited lactase fermentation of unabsorbed sugars infant complains of abdominal beginning of eating, lactose free
deficiency) by intestinal bacteria. distention and colic. diet.
Starts later with addition of cane
The gases produced from Lactose free milk. Then after
sugar to the diet. The infant
Inherited sucrase deficiency fermentation of unabsorbed sugars beginning of eating, sucrose free
complains of abdominal distention
by intestinal bacteria. diet.
and colic.
Accumulation of pyruvate that is
Lactic acidosis Pyruvate dehydrogenase (PDH) converted to lactate lactic
acidosis.
Deficiency of glycolytic enzyme:
- 95%: Pyruvate kinase.
Hemolytic anemia - 4%: Phospho-hexose isomerase.
N.B. RBCs dependent on glycolysis
only for production of energy.
- Avoid: fava beans and certain
Decreased concentration of NADPH drugs that produce H2O2, like:
Glucose 6-phosphate decreased capacity of RBC’s to premaquine, aspirin, and
Favism
dehydrogenase (G6PD). protect itself from oxidative sulfonamides.
damage. - Blood transfusion during the attack
of hemolysis.
Genetic deficiency of L-xylulose Accumulation of L-xylulose and its
Essential pentosuria
reductase excretion in urine.
- Accumulation of G6P & glycogen in
Type I: Von Genetic deficiency of Glucose 6 liver & kidneys fasting
Gierke’s phosphatase (G6Pase) hypoglycemia, hyper-lipidemia,
ketosis, hyperuricemia or Gout.
Glycogen
- Accumulation of glycogen in
storage
muscles painful cramps.
diseases
Type V: - Accumulation of glycogen &
(Glycogenosis) Genetic deficiency of Muscle
McArdle’s decreased ATP increase serum
phosphorylase
Syndrome levels of muscle enzymes (creatine
phosphate, lactate
dehydrogenase).
- Accumulation of F1,6 BP.
- Depletion of liver phosphate &
Fructose 1,6 inhibition of glycogen
Fructose 1,6 Bisphosphatase
Bisphosphatase (F1,6BP) phosphorylase.
(F1,6BPase)
Deficiency - Inhibition of glycogenolysis &
impaired gluconeogenesis.
Fasting hypoglycemia (in infants).
Essential Fructosemia & fructosuria after
Fructokinase enzyme
fructosuria ingestion of fructose or sucrose.
Hereditary
Accumulation of Fructose 1
defects in
Hereditary phosphate (F1P) + depletion of liver
fructose
fructose Aldolase B (liver enzyme) phosphate inhibition of glycogen
metabolism
intolerance phosphorylase hypoglycemia
after fructose or sucrose feeding.
- In eyes: blood galactose reduced
by aldose reductase to form
galactitol (dulcitol) & its - Galactose free diet.
accumulation lead to cataract. - Children can form UDP-Gal from
- Accumulation of galactose 1 UDP-Glc by the epimerase, which
Galactokinase. phosphate & depletion of liver explain their normal growth.
Galactosemia Uridyl transferase (most common). phosphate inhibition of - Later on: children can utilize
4 epimerase. glycogen phosphorylase galactose normally duo to
hypoglycemia after galactose or development of UDP-Gal
lactose feeding. phosphorylase which can replace
- Later on: Liver failure, mental the Gal-1-P Uridyl-transferase.
deterioration & neuronal
dysfunction.
Accumulation of GAGs in tissues
Genetic deficiency of one of the This disease is usually progressive
Cloudy corneas, mental retardation,
Muco-poly-saccharidosis lysosomal enzymes that degrade and leads to death in early
stiff joints & hepato-spleeno-
GAGs childhood.
megaly.
By: Omar Aldurini
2. Diseases of Carbohydrate Metabolism (summary) 2/2
Symptoms &
Disease Definition Types Causes
manifestations
Hyperactivity, hyperplasia or tumors of
pancreatic B-cells (insulinoma).
Hyper-insulism
Over dosage of insulin, even in diabetic
patients, may produce hypoglycemia.
Hypo-function of pituitary, adrenals and
thyroid glands.
Hypo-secretion of anti-
In all these conditions, insulin acts
insulin hormones
unopposed and causes lowering of blood
Fasting glucose.
hypoglycemia Decreased glycogen storage and impaired
- Rapid pulse. Liver diseases
gluconeogenesis.
- Sweating.
Chronic renal disease Impaired gluconeogenesis.
- Headache.
- Von Gierke’s: duo to deficiency of G6Pase.
- Drowsiness.
- F1, 6 BPase deficiency.
It is drop of blood - Tremors.
Hereditary metabolic - Genetic defects that produce impairment
glucose level - If not treated
disorders of fatty acid oxidation. It is explained that
Hypoglycemia below the normal the condition
FA oxidation spares glucose oxidation and
fasting levels may lead to
stimulate gluconeogenesis during fasting.
(>45mg/dl). coma and
Gastrectomy Rapid absorption of glucose
even death Alimentary post-
Rapid rise in blood levels excessive
duo to prandial hypoglycemia
secretion of insulin hypoglycemia.
affection of
Normally: Rise in blood glucose levels
brain tissue.
Secretion of insulin Temporary (short
time) hypoglycemia to fasting level or lower.
Postprandial Reactive hypoglycemia This is known as Reactive Hypoglycemia.
hypoglycemia If prolonged it indicates either exaggerated
insulin response or decreased activity of
anti-insulin hormones.
- Hereditary fructose intolerance: deficiency
Hereditary metabolic of aldolase B.
disorder - Galactosemia: deficiency of Gal-1-P Uridyl-
transferase.
Hyperglycemic Diabetes mellitus Diabetes mellitus.
glucosuria
Occurs when As in emotional, stress glucosuria or
It is the presence blood glucose Epinephrine glucosuria pheochromocytoma (epinephrine secreting
of glucose on level exceeds the tumor).
urine in amounts renal threshold Increase glucose absorption duo to
more than 30 The presence (180 mg/dl) Alimentary glucosuria
Gastrectomy.
mg/dl which are of glucose on
Glucosuria detectable by urine in Congenital glucosuria
Congenital defects on renal tubular
ordinary test amounts more (benign glucosuria or
Normoglycemic or mechanism for reabsorption of glucose.
methods: than 30 mg/dl diabetes innocens)
renal glucosuria
Fehling’s, Acquired renal
In these cases As in case of nephritis.
Benedict’s & diseases
the blood
Urinary strips. It appears during pregnancy and disappears
glucose level is
after labor.
within the
Pregnancy glucosuria N.B. Phlorhizin produces this type of
normal range
glucosuria duo to inhibition of the sodium
dependent transporter of renal tubules.
By: Omar Aldurini