
Recommended
More Related Content
What's hot
What's hot (20)
Similar to Enfermedad de algeman y otros
Similar to Enfermedad de algeman y otros (20)
Enfermedad de algeman y otros
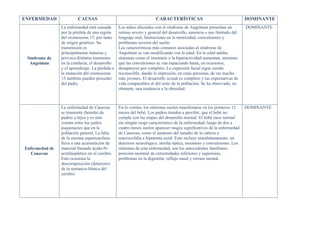
- 1. ENFERMEDAD CAUSAS CARACTERÍSTICAS DOMINANTE Síndrome de Angelman La enfermedad está causada por la pérdida de una región del cromosoma 15, por tanto de origen genético. Su transmisión es principalmente materna y provoca distintos trastornos en la conducta, el desarrollo y el aprendizaje. La pérdida o la mutación del cromosoma 15 también pueden proceder del padre. Los niños afectados con el síndrome de Angelman presentan un retraso severo y general del desarrollo, ausencia o uso limitado del lenguaje oral, limitaciones en la motricidad, convulsiones y problemas severos del sueño. Las características más comunes asociadas al síndrome de Angelman se van modificando con la edad. En la edad adulta, síntomas como el insomnio o la hiperactividad aumentan, mientras que las convulsiones se van espaciando hasta, en ocasiones, desaparecer por completo. La expresión facial sigue siendo reconocible, dando la impresión, en estas personas, de ser mucho más jóvenes. El desarrollo sexual es completo y las expectativas de vida comparables al del resto de la población. Se ha observado, no obstante, una tendencia a la obesidad. DOMINANTE Enfermedad de Canavan La enfermedad de Canavan se transmite (hereda) de padres a hijos y es más común entre los judíos asquenacíes que en la población general. La falta de la enzima aspartoacilasa lleva a una acumulación de material llamado ácido-N- acetilaspártico en el cerebro. Esto ocasiona la descomposición (deterioro) de la sustancia blanca del cerebro. En lo común, los síntomas suelen manifestarse en los primeros 12 meses del bebé. Los padres tienden a percibir, que el bebé no cumple con las etapas del desarrollo normal. El bebé nace normal sin ningún rasgo característico de la enfermedad, luego de dos a cuatro meses suelen aparecer rasgos significativos de la enfermedad de Canavan, como el aumento del tamaño de la cabeza o macrocefalia e hipotonía axial. Esto incluye simultáneamente, un deterioro neurológico, atrofia óptica, insomnio y convulsiones. Los síntomas de esta enfermedad, son los antecedentes familiares, posición anormal de extremidades inferiores y superiores, problemas en la digestión, reflujo nasal y retraso mental. DOMINANTE
- 2. Síndrome de Charcot–Marie– Tooth La enfermedad de Charcot- Marie-Tooth es uno de los trastornos más comunes relacionados con los nervios que se transmiten de padres a hijos (hereditarios). Problemas en al menos 40 genes causan diferentes formas de esta enfermedad. La enfermedad lleva a daño o destrucción de la cubierta (vaina de mielina) alrededor de las fibras nerviosas. La neuropatía de la enfermedad de CMT afecta los nervios motores y sensoriales. Una característica típica incluye debilitaciones en los pies y de los músculos inferiores de la pierna, que pueden dar lugar a una deformación del pie y generar una marcha a pasos grandes que desencadena en tropiezos o caídas frecuentes. Las deformidades del pie, tales como arcos altos y dedos en martillo (una condición en la cual la coyuntura central de un dedo del pie se dobla hacia arriba) son también características debido a la debilidad de los músculos más pequeños del pie. Además, la parte inferior de las piernas puede adquirir un aspecto "de botella de champán invertida" debido a la pérdida de masa muscular. Conforme progresa la enfermedad, pueden ocurrir debilidades y atrofias musculares en las manos, dando como resultado dificultades en las capacidades motoras. Aunque los nervios sensitivos también están involucrados, muy raras veces los pacientes sufren de entumecimientos o dolores significativos. DOMINANTE Daltonismo El daltonismo ocurre cuando hay un problema con los gránulos (pigmentos) que perciben el color en ciertas neuronas del ojo, llamadas conos. Estas células se encuentran en la retina, la capa de tejido sensible a la luz que recubre la parte posterior del ojo. Si sólo falta un pigmento, usted puede tener dificultad para diferenciar entre el rojo y el verde, que es el tipo más común de daltonismo. Si falta un pigmento diferente, usted puede tener dificultad para ver los colores azul y amarillo. Las personas con daltonismo para los colores Es un defecto genético que consiste en la imposibilidad de distinguir los colores (discromatopsia). Aunque ningún daltónico confunde los mismos colores que otros, incluso pertenecientes a la misma familia, es muy frecuente que confundan el verde y el rojo; sin embargo, pueden ver más matices del violeta que las personas de visión normal y son capaces de distinguir objetos camuflados. También hay casos en los que la incidencia de la luz puede hacer que varíe el color que ve el daltónico. NO DOMINANTE
- 3. azul y amarillo generalmente tienen problemas para identificar también los colores rojos y verdes. Síndrome de Down El Síndrome de Down está causado por un número mayor de cromosomas. Normalmente, hay 23 pares de cromosomas. Una mitad proviene de la madre y la otra del padre. El Síndrome de Down no lo causa algo que hacen la madre o el padre antes de que nazca el niño. Cualquiera puede tener un niño con Síndrome de Down. Pero cuanto mayor sea la madre, mayor es el riesgo de tener un bebé con Síndrome de Down. Una persona con el síndrome puede tener algunas o todas de las siguientes condiciones físicas: ojos de almendra, un solo pliegue palmar transversal (también conocido como pliegue simiesco), dedos curtinhos, fisuras palpebrales, puente nasal plano, lengua que sobresale (debido a la cavidad oral pequeña ), cuello corto, manchas blancas en el iris conocidas como manchas Brushfield, demasiada flexibilidad en las articulaciones, los defectos congénitos del corazón , el espacio excesivo entre el dedo gordo y el segundo dedo. A pesar de la apariencia a veces común entre las personas con síndrome de Down, hay que recordar que lo que realmente caracteriza al individuo es su familia genética, lo que hace que sean como sus padres y hermanos. Los niños con síndrome de Down tienen una desventaja frente a diversos niños sin el síndrome, ya que la mayoría de las personas con síndrome de Down tienen retraso mental desde leve ( CI 50-70) a moderado (CI 35 -50) , con el coeficiente intelectual de los niños con síndrome de Down mosaico de tipo típicamente 10-30 puntos más alto. Además, las personas con síndrome de Down pueden tener anormalidades graves que afectan a cualquier sistema del cuerpo . Otra característica es la frecuente microcefalia , un reducido peso y tamaño de la cerebro . El progreso en el aprendizaje es también típicamente afectadas por enfermedades y discapacidades físicas, tales como las enfermedades infecciosas recurrentes problemas en el corazón , problemas con la visión ( miopía , astigmatismo o estrabismo ) y la audición . DOMINANTE
- 4. Síndrome de Edwards El síndrome de Edwards es una anomalía cromosómica caracterizada por la presencia de una copia adicional de material genético del cromosoma 18, tanto si esta información es un cromosoma entero (hablaríamos entonces de una trisomía 18), como si es parcial (como una translocación). Los efectos del exceso cromosómico variarán en función de esto último, aparte del historial genético (background) y del azar. Crecimiento lento. Presentan retaso mental. Puños cerrados. El 80% de los nacidos son mujeres. Tienen el segundo y quinto dedo superpuestos al tercero y cuarto. Pies con arco plantar escaso. Talones salientes. Cráneo en forma de fresa. Quistes en plexo coroideo. Hendiduras faciales. Agenesia del cuerpo calloso. Defectos renales. Edema nucal. Acortamiento de extremidades. Mielomeningocele: espina bífida con hernia en médula espinal y sus meninges. Malformaciones cardíacas. Suelen morir por insuficiencia cardiaca o por neumonía. Tiene tasa de letalidad intrauterina alta. NO DOMINANTE
- 5. Espina bífida Habitualmente la espina bífida proviene de la unión de una predisposición genética y factores ambientales. Entre las causas ambientales podemos señalar: El 98% de los casos se debe a un déficit de folatos en la madre en los momentos previos o inmediatamente posteriores a producirse el embarazo. Tratamiento materno con fármacos: ácido valproico (anticonvulsionante), etetrinato (tratamiento para la psoriasis y el acné), carbamazepina (tratamiento psiquiátrico) y medicamentos hormonales. Conductuales: Parálisis y pérdida de sensibilidad por debajo del nivel de la lesión. Debilidad por debajo de la lesión. Problemas de habilidad manipulativa. Problemas emocionales. Cognitivas: Escritura. Matemáticas. Plástica y dibujo. Problemas Visuales. Problema de orientación. Problemas de laterización. Socio relacionales: Auto concepto y autoestima. Dificultades de relación. Aislamiento Social. Interacción Significativa. NO DOMINANTE
- 6. Fenilcetonuria Afecta a la manera en que el cuerpo procesa los alimentos: los niños afectos no pueden procesar un componente de las proteínas (aminoácido) llamado fenilalanina por falta del enzima correspondiente. Como resultado la fenilalanina se acumula en sangre y provoca daños cerebrales y retraso mental. Se hereda de forma recesiva: Ambos padres deben tener el gen defectuoso, La probabilidad de que se lo pasen a sus hijos es del 75 %, La probabilidad de que el hijo sea completamente normal (de que lleve dos copias normales del gen, y por tanto ni padezca la enfermedad ni pueda transmitirla) es del 25 %. Cifosis. Pies planos. Espina bífida. Sindactilia en los dedos de los pies. Bloqueo cardiaco intraventricular. Hipogenitalismo. Dermografismo. Sensibilidad a la luz. Hipersegmentación de las células neutrófilas de la sangre. Disminución de la tolerancia a la galactosa. Metabolismo basal ligeramente elevado. Las etapas del desarrollo habitual, la edad en la que el niño se sienta y habla, a veces, se alcanzan a la edad normal, pero, de ordinario, se retrasa. En la edad límite en que debe esperarse que el niño normalmente realice estos actos, el 35% no puede andar y el 63% no puede hablar. Estos niños, en general, tienen un peso y talla promedio por debajo del correspondiente a su edad. En la mitad de los casos tiene microcefalia y prominencia del maxilar. Sus movimientos son lentos y patosos y a menudo suelen adoptar la posición de sastre. Las anomalías del tono muscular que contribuyen a estos cambios son de origen neurológico.2 de cada 3 pacientes tienen hiperreflexiatediciosa e hipercinesia sobreañadida estos últimos son voluntarios y muy variados. DOMINANTE
- 7. Fibrosis quística La fibrosis quística (FQ) es causada por un gen defectuoso que lleva al cuerpo a producir un líquido anormalmente espeso y pegajoso llamado moco. Este moco se acumula en las vías respiratorias de los pulmones y en el páncreas, el órgano que ayuda a descomponer y absorber los alimentos. Esta acumulación de moco pegajoso ocasiona infecciones pulmonares potencialmente mortales y serios problemas digestivos. Esta enfermedad también puede afectar las glándulas sudoríparas y el aparato reproductor masculino. La sintomatología de la fibrosis quística varía en función de la edad del individuo, el grado en que se ven afectados órganos específicos, la terapéutica instituida previamente, y los tipos de infecciones asociadas. Esta enfermedad compromete al organismo en su totalidad y muestra su impacto sobre el crecimiento, la función respiratoria, la digestión. El periodo neonatal se caracteriza por un pobre aumento de peso y por obstrucción intestinal producida por heces densas y voluminosas. Otros síntomas aparecen, más tarde, durante la niñez y al inicio de la adultez. Éstos incluyen retardo del crecimiento, advenimiento de la enfermedad pulmonar, y dificultades crecientes por la malabsorción de vitaminas y nutrientes en el tracto gastrointestinal. A la mayoría de los niños se les diagnostica fibrosis quística antes del primer año de vida, cuando la mucosidad pegajosa que afecta pulmones y páncreas, comienza a mostrar su impacto. En el tracto respiratorio, esas secreciones sirven como caldo de cultivo para diversas bacterias responsables de infecciones crónicas, con deterioro progresivo y permanente del parénquima pulmonar. Conforme se agrava la condición respiratoria, los pacientes sufren hipertensión pulmonar. Por otra parte, en el páncreas, el moco obstruye el tránsito de las enzimas sintetizadas por la glándula e impide que lleguen hasta el intestino para digerir y absorber el alimento. DOMINANTE
- 8. Hemofilia Las causas de la hemofilia se encuentran en la ausencia, deficiencia o conformación inadecuada de determinadas proteínas que forman parte de la denominada cascada de coagulación. Cuando se produce una herida, el organismo da la orden de movilizar una serie de componentes presentes en la sangre (que se encuentran en circulación en todo momento, haya o no herida) que acudirán al punto lesionado, formando un muro que impide la salida de la sangre. La formación de este “muro” (coágulo) sucede tras la actuación de unas proteínas denominadas factores de coagulación. El proceso mediante el cual estos activan el mecanismo de coagulación recibe el nombre de cascada de coagulación, ya que cada uno de ellos (12 en total) activa al siguiente. Debido a esta activación en cascada, si uno de los factores no se encuentra presente, o lo está de manera deficiente, será la causa de un fallo en todo el proceso. Se caracteriza por la aparición de hemorragias internas y externas debido a la deficiencia parcial de una proteína coagulante denominada globulina anti hemofílica (factor de coagulación). Los factores de coagulación son un grupo de proteínas responsables de activar el proceso de coagulación. Hay identificados 12 factores ( I, II, ..., XIII. El factor VI no ha sido asignado). Los factores de coagulación actúan en cascada, es decir, uno activa al siguiente; si se es deficitario de un factor, no se produce la coagulación o se retrasa mucho Cuando hay carencia o déficit de algún factor de coagulación, la sangre tarda más tiempo en formar el coágulo y, aunque llegue a formarse, no es consistente y no se forma un buen tapón para detener la hemorragia, por tanto, en los hemofílicos graves, incluso pequeñas heridas pueden originar abundantes y hasta mortales pérdidas de sangre. La agregación plaquetaria no está alterada en los hemofílicos (el tiempo de sangrado de Ivy es normal). También es patognomónico el hemartrosis (sangrado interno de las articulaciones), pues en ellas no se encuentra factor tisular, que es el desencadenante de la vía extrínseca de la coagulación, la única funcionante en hemofílicos. Hay tres variedades de hemofilia: la A, cuando hay un déficit del factor VIII de coagulación, la B, cuando hay un déficit del factor IX de coagulación, y la C, que es el déficit de factor XI. DOMINANTE
- 9. Síndrome de Ehlers-Danlos Existen seis grandes tipos y al menos cinco tipos menores del síndrome de Ehlers- Danlos (SED). Una variedad de mutaciones (cambios) genéticas causa problemas con el colágeno, el material que brinda estructura y fortaleza a la piel y el hueso, los vasos sanguíneos y los órganos internos. El colágeno anormal lleva a síntomas asociados con el síndrome de Ehlers-Danlos. En algunas formas de la enfermedad, esto puede incluir ruptura de órganos internos o válvulas cardíacas anormales. Los antecedentes familiares son un factor de riesgo en algunos casos. Dolor de espalda. Hiperlaxitudligamentaria. Piel que se estira, presenta equimosis y se daña fácilmente. Cicatrización fácil y curación de heridas deficiente. Pies planos. Aumento de la movilidad articular, crujido en las articulaciones, artritis temprana. Dislocación articular. Dolor articular. Ruptura prematura de membranas durante el parto. Piel muy suave y aterciopelada. Problemas de visión. DOMINANTE
- 10. Hipermovilidad Los síntomas asociados a la hiperlaxitud articular se definen como "síndrome de hiperlaxitud articular". Según los especialistas, solamente un bajo porcentaje de las persona con hiperlaxitud presentan síntomas, llegando al 10% las que presentan problemas a nivel de aparato locomotor. Por ello, es necesario diferenciar entre hiperlaxitud articular (hiperlaxitud sin síntomas) y padecer un síndrome de hiperlaxitud articular (hiperlaxitud articular con síntomas) En el caso del síndrome, sus causas todavía no están claras, aunque sí está probado que son de carácter genético. La causa más probable es una mutación genética que provoca que las fibras de colágeno repercutan en la elasticidad y fragilidad de ligamentos, tendones, vasos sanguíneos, piel, etc. Al aumentar la elasticidad de las mismas, la posibilidad de sufrir lesiones es mayor. Dolor en músculos y articulaciones (tanto en las superiores como inferiores como axiales, es decir, muñecas, dedos, codos, hombros, cervicales, espalda, caderas, rodillas, tobillos...), rigidez muscular (espasmos). Enfermedades con los tejidos blandos, tales como tendinitis, capsulitis, torceduras de tobillo, torticolis, luxaciones (huesos que se salen de su sitio). Enfermedades ligadas a la columna, tales como la lumbalgia, la escoliosis o el pie plano. Síntomas fuera de las articulaciones ligadas a la hiperlaxitud: Aumento de la distensibilidad de la piel, varices, hernias. DOMINANTE
- 11. Síndrome de Joubert Las causas del Síndrome de Joubert no son muy claras, pero se dice que la causa es por un gen, por lo que se suma el hecho, de una deleción de un cromosoma muy pequeño o nulo, pero no se sabe con certeza sobre la causa de este síndrome. Se sabe muy poco acerca de cuál o cuáles son los genes que desencadenan la aparición del síndrome. Algunas investigaciones suman la idea que puede ser un gen encargado de proteínas asociadas con el desarrollo de vellosidades de celular del cerebelo. La herencia de este síndrome es autosómica y recesiva. Principalmente se caracteriza por presentar hipotonía o pérdida del tono muscular, hiperpnea (a veces acompañada de apnea del sueño), ataxia troncal, displasia y el movimiento anormal de los ojos. Todo esto, unido a la falta de vermis en el cerebelo provoca un retraso general en el desarrollo, fatiga y la aparición de retrasos mentales de distinto grado. Pueden darse casos de movimientos imitativos (especulares). A veces aparecen otro tipo de anomalías menos comunes, como la hipersensibilidad al ruido, polidactilia, meningoencefaloceles, quistes renales o microcefalia. Este síndrome se ha asociado muy a menudo con el autismo, ya que muchos pacientes diagnosticados muestran criterios para designarlos como pacientes de autismo. Lo más importante es que los síntomas sólo se muestran en los primeros años del paciente. La persona puede nacer con el trastorno genético, pero si no muestra los síntomas a los pocos meses o años, la enfermedad no remitirá y será un individuo sano. Si por el contrario los síntomas permanecen, podrá tratarse, pero la afección será definitiva. NO DOMINANTE
- 12. Síndrome de Klinefelter La mayoría de las personas tienen 46 cromosomas. Los cromosomas contienen todos los genes y el ADN, los pilares fundamentales del cuerpo. Dos cromosomas sexuales determinan si usted se convertirá en niño o en niña. Las mujeres normalmente tienen dos cromosomas sexuales XX, mientras que los hombres normalmente tienen un cromosoma X y un cromosoma Y. El síndrome de Klinefelter se presenta cuando un niño varón nace con al menos un cromosoma X extra. Por lo regular, esto ocurre debido a un cromosoma X adicional. Esto se escribiría como XXY. Proporciones corporales anormales (piernas largas, tronco corto, hombro igual al tamaño de la cadera). Agrandamiento anormal de las mamas (ginecomastia). Infertilidad. Problemas sexuales. Vello púbico, axilar y facial menor a la cantidad normal. Testículos pequeños y firmes. Estatura alta. DOMINANTE
- 13. Neurofibromatosis La neurofibromatosis(NF1) es una enfermedad hereditaria. Si cualquiera de los padres la padece, cada uno de sus hijos tiene un 50% de probabilidades de tener la enfermedad. La NF1 también aparece en familias sin antecedentes previos de la afección. En estos casos, es causada por un cambio nuevo en un gen (mutación) en el espermatozoide o en el óvulo. La NF1 es ocasionada por problemas con un gen para una proteína llamada neurofibromina. La neurofibromatosis causa crecimiento incontrolado de tejido en los nervios, lo cual puede ejercer presión sobre los nervios afectados. Esto puede causar dolor, daño nervioso grave y pérdida de la función en el área estimulada por dicho nervio. Se pueden presentar problemas con la sensibilidad o el movimiento, según cuáles sean los nervios afectados. La enfermedad puede diferir mucho de una persona a otra, incluso entre personas de la misma familia que tengan el gen para NF1. Las manchas de color café con leche son el síntoma distintivo de la neurofibromatosis. Muchas personas tienen 1 o 2 manchas pequeñas de color café con leche; sin embargo, los adultos con 6 o más manchas superiores a 1.5 cm (0.5 cm en los niños) de diámetro tienen probabilidad de padecer neurofibromatosis. En la mayoría de las personas con esta afección, estas manchas pueden ser el único síntoma. DOMINANTE
- 14. Enfermedad de Pelizaeus- Merzbacher La enfermedad de PelizaeusMerzbacher es una afección hereditaria del Sistema Nervioso Central, que se particulariza por la pérdida avanzada de la grasa de la vaina de mielina que está por encima de las fibras nerviosas del cerebro y las glándulas adrenales. La enfermedad de PelizaeusMerzbacheresta clasificada como una leucodistrofiasudanófila tipo recesivo. Los síntomas de la enfermedad de PelizaeusMerzbacher son espasmos en los ojos y movimientos de cabeza, acompañado de parálisis, contracción muscular, movimientos sin coordinación, demencia, convulsiones y atrofia del nervio óptico. En los primeros meses de vida, la enfermedad de PelizaeusMerzbacher se caracteriza por presentar ciertos síntomas como disminución del tono muscular, ojos sacudidos, movimientos anormales y frecuentes de la cabeza y de los miembros, lo que dificulta al paciente desde muy niño, poder hablar y sentarse bien. La afectación a los pacientes con la enfermedad de PelizaeusMerzbacher varía en cada persona, hay casos de niños que mueren antes de los 10 años, en otras situaciones suele presentar durante el resto de su vida, dificultades para caminar y cierta inestabilidad. DOMINANTE
- 15. Síndrome de Patau La mayoría de los casos de síndrome de Patau se deben a una trisomía del cromosoma 13 (consecuencia de una no disyunción meiótica, principalmente en el gameto materno). Aproximadamente un 20% de casos se deben a traslocaciones, siendo la t(13q14) la más frecuente. Sólo un 5% de dichas traslocaciones es heredada de uno de los progenitores. En el caso de la traslocación, aunque los padres estén sanos tienen posibilidad de pasar la enfermedad a su descendencia. Los mosaicos representan 5% de los casos de trisomía 13. Anomalías en el sistema nervioso Retraso mental Dilatación de la bifurcación ventricular Alargamiento del surco posterior Aumento de tamaño del riñón Anomalías cardíacas Comunicación interventricular Displasia valvular Tetralogía de Fallot Anomalías de miembros Polidactilia Pie valgo Anomalías en abdomen Onfalocele Extrofía vesicular Hipotonía muscular DOMINANTE
- 16. Síndrome de Prader-Willi El síndrome de Prader-Willi es causado por la carencia de un gen en parte del cromosoma 15. Normalmente, cada uno de los padres transmite una copia de este cromosoma. La mayoría de los pacientes con este síndrome carecen del material genético en parte del cromosoma del padre. El resto de los pacientes con frecuencia tiene dos copias del cromosoma 15 de la madre. Los cambios genéticos ocurren en forma aleatoria. Los pacientes generalmente no tienen antecedentes familiares de esta afección. Los signos del síndrome de Prader-Willi pueden verse al nacer. Los recién nacidos suelen ser pequeños y flácidos. Los recién nacidos varones pueden tener criptorquidia. Otras características pueden ser: Problemas para comer durante la lactancia que llevan a un aumento de peso deficiente. Ojos en forma de almendra. Desarrollo motor retardado. Cráneo bifrontal estrecho. Aumento rápido de peso. Estatura corta. Desarrollo mental lento. Manos y pies muy pequeños. Los niños presentan una ansiedad intensa por la comida y harán casi cualquier cosa por obtenerla, lo cual puede producir un aumento de peso incontrolable y obesidad mórbida. Dicha obesidad puede llevar a que se presente diabetes tipo 2, hipertensión arterial y problemas pulmonares y articulares. NO DOMINANTE
- 17. Enfermedad de Tay-Sachs La enfermedad de Tay-Sachs es causada por un gen defectuoso en el cromosoma 15. Cuando ambos padres portan el gen defectuoso para esta enfermedad, el hijo tiene un 25% de probabilidades de desarrollarla. El niño tiene que recibir dos copias del gen defectuoso, una de cada uno de los padres, para resultar enfermo. Si sólo uno de los padres le transmite dicho gen defectuoso, el niño se denomina portador y no se enfermará, pero tendrá el potencial de transmitirles la enfermedad a sus hijos. Sordera Disminución en el contacto visual, ceguera Disminución del tono muscular (pérdida de la fuerza muscular) Retraso en el desarrollo de habilidades mentales y sociales Demencia Aumento del reflejo de sobresalto Irritabilidad Apatía o desgano Pérdida de las destrezas motrices Parálisis o pérdida de la función muscular Crisis epiléptica Crecimiento lento DOMINANTE
- 18. Síndrome de Turner La cantidad normal de cromosomas humanos es 46. Los cromosomas contienen todos los genes y el ADN, los pilares fundamentales del cuerpo. Dos de estos cromosomas, los sexuales, determinan si uno ha de ser hombre o mujer. Las mujeres normalmente tienen dos de los mismos cromosomas sexuales, que se escriben como XX, mientras que los hombres tienen un cromosoma X y un cromosoma Y, que se escriben como XY. En el síndrome de Turner, el cual sólo ocurre en las mujeres, a las células les falta todo o parte de un cromosoma X. Lo más común es que la paciente femenina tenga sólo un cromosoma X; mientras que otras pueden tener dos cromosomas X, pero uno de ellos está incompleto. Algunas veces, una mujer tiene algunas células con los dos cromosomas X, pero otras células tienen sólo uno. Los posibles síntomas en los bebés pequeños abarcan: Manos y pies hinchados. Cuello ancho y unido por membranas. En las niñas mayores, se puede observar una combinación de los siguientes síntomas: Desarrollo retrasado o incompleto en la pubertad, que incluye mamas pequeñas y vello púbico disperso. Tórax plano y ancho en forma de escudo. Párpados caídos. Ojos resecos. Infertilidad. Ausencia de períodos (ausencia de la menstruación). Resequedad vaginal, que puede llevar a relaciones sexuales dolorosas. DOMINANTE